Каталог
Вернуться к результатам поиска
Unipro UGENE

Вернуться к результатам поиска
Unipro UGENE
Unipro UGENE предоставляет множество методов обработки генетических и белковых последовательностей. Это — анализ, аннотирование, сравнение, поиск, выравнивание, моделирование, комплексные вычисления. Разнообразная визуализация, навигация и масштабирование, поддержка множества генетических форматов данных , оптимизация алгоритмов, возможности редактирования и графического построения различных вычислительных схем существенно ускоряют работу. UGENE отлично работает на Windows, Mac OS, Linux и прост в установке и использовании. Обладает интерфейсом как на русском, так и на английском языке.
Ниже представлены основные возможности продукта:
Создание, редактирование и аннотирование нуклеотидных и белковых последовательностей.
Быстрый поиск в последовательности
Множественное выравнивание последовательностей: ClustalW, ClustalO, MUSCLE, Kalign, MAFFT, T-Coffee
Поиск в онлайн базах данных: NCBI, PDB, UniProtKB/Swiss-Prot, UniProtKB/TrEMBL, серверы DAS
Онлайн и локальный BLAST поиск
ПЦР in silico
Поиск открытых рамок считывания
Рестрикционный анализ со встроенной базой данных ферментов рестрикции REBASE
Интегрированный пакет Primer3 для дизайна ПЦР праймеров
Аннотирование плазмид
Клонирование in silico
Выравнивание на геном с помощью Bowtie, BWA или UGENE Genome Aligner
Визуализация выравненных коротких прочтений с помощью UGENE Assembly Browser
Поиск геномных вариаций с помощью SAMtools
Обработка сырых данных NGS
Анализ RNA-Seq данных с помощью TopHat и инструментов Cufflinks
SPAdes de novo ассемблер
Поиск гомологов с HMMER2 и HMMER3
Работа с хроматограммами
Поиск сайтов связывания транскрипционных факторов с использованием весовых матриц или алгоритма SITECON
Поиск повторов в последовательности ДНК: прямых, обратных, тандемных
Локальное выравнивание последовательности с использованием оптимизированной версии алгоритма Смита-Ватермана
Построение филогенетических деревьев (с помощью IQ-TREE, PHYLIP Neighbor Joining, MrBayes или PhyML Maximum Likelyhood) и редактирование деревьев
Комбинирование различных алгоритмов в вычислительную схему с помощью дизайнера вычислительных схем
Сборки контигов (CAP3)
Отображение 3D структуры белков для форматов PDB и MMDB formats, поддержка стереоэффекта
Предсказание вторичной структуры белка с помощью алгоритмов GOR IV и PSIPRED
Конструирование точечных графиков для ДНК последовательностей
Выравнивание мРНК (Spidey)
Поиск шаблона результатов различных алгоритмов в нуклеотидной последовательности с помощью дизайнера запросов.
Ниже представлены основные возможности продукта:
Создание, редактирование и аннотирование нуклеотидных и белковых последовательностей.
Быстрый поиск в последовательности
Множественное выравнивание последовательностей: ClustalW, ClustalO, MUSCLE, Kalign, MAFFT, T-Coffee
Поиск в онлайн базах данных: NCBI, PDB, UniProtKB/Swiss-Prot, UniProtKB/TrEMBL, серверы DAS
Онлайн и локальный BLAST поиск
ПЦР in silico
Поиск открытых рамок считывания
Рестрикционный анализ со встроенной базой данных ферментов рестрикции REBASE
Интегрированный пакет Primer3 для дизайна ПЦР праймеров
Аннотирование плазмид
Клонирование in silico
Выравнивание на геном с помощью Bowtie, BWA или UGENE Genome Aligner
Визуализация выравненных коротких прочтений с помощью UGENE Assembly Browser
Поиск геномных вариаций с помощью SAMtools
Обработка сырых данных NGS
Анализ RNA-Seq данных с помощью TopHat и инструментов Cufflinks
SPAdes de novo ассемблер
Поиск гомологов с HMMER2 и HMMER3
Работа с хроматограммами
Поиск сайтов связывания транскрипционных факторов с использованием весовых матриц или алгоритма SITECON
Поиск повторов в последовательности ДНК: прямых, обратных, тандемных
Локальное выравнивание последовательности с использованием оптимизированной версии алгоритма Смита-Ватермана
Построение филогенетических деревьев (с помощью IQ-TREE, PHYLIP Neighbor Joining, MrBayes или PhyML Maximum Likelyhood) и редактирование деревьев
Комбинирование различных алгоритмов в вычислительную схему с помощью дизайнера вычислительных схем
Сборки контигов (CAP3)
Отображение 3D структуры белков для форматов PDB и MMDB formats, поддержка стереоэффекта
Предсказание вторичной структуры белка с помощью алгоритмов GOR IV и PSIPRED
Конструирование точечных графиков для ДНК последовательностей
Выравнивание мРНК (Spidey)
Поиск шаблона результатов различных алгоритмов в нуклеотидной последовательности с помощью дизайнера запросов.

Гаплоидный эволюционный конструктор
Программа позволяет конструировать и анализировать компьютерные модели микробных сообществ с учётом следующих уровней биологической организации: генетического, метаболического, организменного, популяционного, экологического. Программа строит модель многовидового микробного сообщества, функционирование клеток каждого вида в котором описывается генной/метаболической сетью, а также процессами транспорта метаболитов в среде обитания (биореакторе).
Произведено в: Новосибирск
Protomenal
Сервис позволяет сканировать последовательность с максимальной длиной 40 000 аминокислот на наличие совпадений с базами данных сигнатур белков.
Novel Software Systems
Новосибирск
Произведено в: Новосибирск

GENOMENAL
Загрузка и обработка данных
Автоматическое распознавание платформ секвенирования и типов данных
Поддержка различных форматов входных файлов: FASTQ, BAM, VCF
Обработка данных происходит в автоматическом режиме и не требует вмешательства
Все недочеты в данных будут выявлены на этапе обработки и продемонстрированы пользователю
Variant Viewer — инструмент для просмотра генетических вариантов
Удобный и интуитивно понятный интерфейс
Гибкость функционала за счет системы продвинутых фильтров
Обработка миллионов генетических вариантов
Аннотация
ClinVar, Ensembl, 1KG, dbNSFP, dbSNP, gnomAD v3, VarSome и другие открытые базы
Интеграция с IGV, PubMed, HPO, Google Scholar
Автоматический поиск по научным публикациям и актуальным рекомендациям профессионального сообщества по различным нозологиям
Возможность использовать собственную базу генетических находок
Novel Software Systems
Новосибирск
Произведено в: Новосибирск
GeneNetStudio
Новая клиентская часть системы GeneNet. Она предоставляет возможности создания, редактирования и дальнейшего анализа генных сетей. GeneNetStudio поддерживает систему отсеков, которые позволяют описывать генную сеть на основе пространственных особенностей. Программный комплекс включает в себя такие подсистемы: (1) поиск схем регулирования; (2) филогенетический распад генной сети; (3) преобразование ассоциативно-семантических сетей молекулярно-генетических взаимодействий из системы ANDCell в генные сети; (4) преобразование сетевых моделей в двудольное представление и другие.
В состав системы GeneNet входят: база данных по компонентам генной сети, Java-программа для визуализации данных.
Разработчики программы: Группа моделирования молекулярно-генетических систем Института цитологии и генетики СО РАН
Произведено в: Новосибирск
Генокарта
Электронная научно-популярная энциклопедия по генетике человека на русском языке. Генокарта содержит статьи о генах человека и генетических полиморфизмах, о болезнях и фенотипах с известной генетической составляющей, словарь терминов и новости, относящиеся к генетике человека, а также более 15 миллионов статей о мутациях и вариациях, автоматически сгенерированных на основании данных из открытых источников: dbSnp, ClinVar, SNPedia, GWAS. Статьи о болезнях и фенотипах написаны экспертами с учетом особенностей российской популяции и российской системы здравоохранения. Пользователи Генокарты — врачи, медицинские генетики, ученые-генетики, студенты, пациенты и их родственники. С 2022 года любой желающий может аннотировать генетические данные на Генокарте.
Novel Software Systems
Новосибирск
Произведено в: Новосибирск

Oncobox Sample Storage
Oncobox Sample Storage является RESTfull сервисом. Для сравнения и полноценного анализа нескольких образцов требуются дополнительные данные о пациенте, терапии и так далее. Часто они доступны только в неупорядоченном или неполном виде. Предварительное структурирование позволяет быстро отбирать для анализа требуемые образцы и сравнивать результаты друг с другом.
ПО может использоваться в:
В научных исследованиях, где требуется анализ групп образцов. Например, при поиске новых молекулярных мишеней, исследовании эффективности препаратов, поиске клинико-патологических зависимостей.
В медицинской деятельности для накопления данных о пациентах и последующего анализа транскриптомных изменений в опухоли для выбора наиболее эффективной терапии.
Онкобокс
Долгопрудный
Произведено в: Долгопрудный

PDBSiteScan
Программа предназначена для функциональной аннотации белковых молекул, реконструкции сетей белок-белковых и белок-лиганд взаимодействий. Программа позволяет распознавать функциональные сайты в пространственных структурах белков, включая: сайты каталитических центров ферментов, сайты посттрансляционных модификаций белков, сайты связывания ионов металлов, сайты связывания органических и неорганических лигандов, сайты связывания ДНК и РНК, а также восстанавливать пространственные структуры комплексов лигандов с активными сайтами белков.
Программа обеспечивает выявление функционально важных свойств структуры белков, необходимых для решения задач идентификации функции белков, поиска мишеней для лекарственных препаратов и целенаправленного конструирования белков с улучшенными медико-биологическими свойствами, а также поиск и конструирование низкомолекулярных лигандов, способных связываться с активными центрами белков.
Автор программы: Иванисенко Владимир Александрович
Произведено в: Новосибирск

ArtSite
База данных ArtSite предназначена для аннотации экспериментальной информации по структуре сайтов связывания транскрипционных факторов (ТФ) у про- и эукариот. База основана на описании структуры сайтов связывания ТФ с помощью частотных матриц, которые построены в результате выравнивания представительных выборок нуклеотидных последовательностей сайтов связывания ТФ.
Выборки сайтов созданы на основе in vitro синтезированных последовательностей, выявленных с помощью различных методов селекции, и описанных в научных статьях, опубликованных в открытой печати. Матрицы были построены на основе выравниваний репрезентативных выборок сайтов связывания транскрипционных факторов, содержащих в общей сложности более 10 тысяч последовательностей.
Язык программирования: Icarus
СУБД: Sequence Retrieval System (SRS)
Разработчики программы (базы данных):
Хлебодарова Тамара Михайловна
Произведено в: Новосибирск
WebProAnalyst
WebProAnalyst — это инструмент анализа, предназначенный для сканирования количественных взаимосвязей структура-активность в семействах белков. Инструмент позволяет пользователям искать корреляции между активностью белка и физико-химическими характеристиками (т.е. гидрофобностью или альфа-спиральной амфипатичностью) в запрашиваемых последовательностях. WebProAnalyst использует выровненные аминокислотные последовательности и данные об активности белков (pK, Km , ED50 и другие ).
Программа позволяет предсказывать мутации для целенаправленного изменения величины активности белков или других их свойств на основе количественного анализа взаимосвязи структура-активность в белковых семействах.
Автор программы Иванисенко Владимир Александрович
Произведено в: Новосибирск
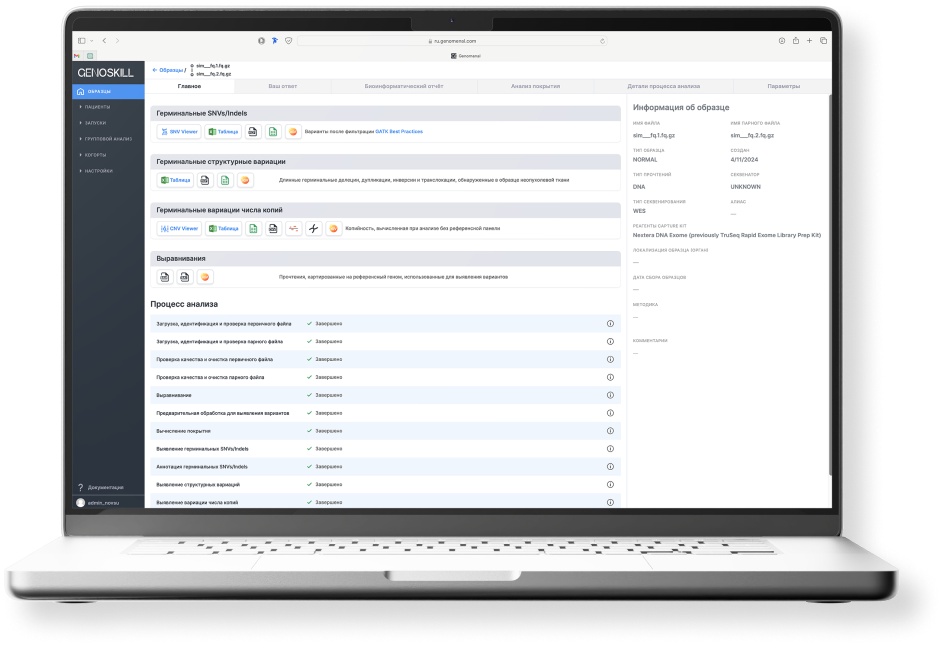
Genoskill
Тренажер GENOSKILL предназначен для студентов, магистрантов, аспирантов, слушателей курсов повышения квалификации и любых других категорий обучающихся. Эта программа предоставит вам возможность углубить знания в области медицинской генетики, биоинформатики и геномики, попрактиковаться в получении навыков и понимании принципов интерпретации генетических вариантов у пациентов с наследственными заболеваниями.
Обучение проходит путем решения клинических задач на синтетических данных.
В образовательную серию вошли клинические случаи с распространенными, известными каждому медицинскому работнику заболеваниями. В базовую серию включены результаты биоинформатической обработки 10 экзомов и задачи поиска причин наследственной патологии с различными типами наследования. Решения предполагают получение компетенций и навыков поиска различных по эффекту влияния на белок патогенных вариантов, комплексных аллелей, инсерций, делеций и даже двойной диагноз, а также другие особенности данных, необходимые для погружения в клиническую интерпретацию.
Novel Software Systems
Новосибирск
Произведено в: Новосибирск

Программное обеспечение для фрагментного анализа Helicon Genmap
Helicon Genmap — это гибкое программное обеспечение (ПО) для анализа данных фрагментного анализа после капиллярного электрофореза. Предназначено для установления размеров ДНК-фрагментов и определения генетического профиля человека, животных и растений. Алгоритм ПО основан на современном математическом анализе, снабжён системой оценки качества и модернизированными инструментами для обзора данных. Имеется возможность создания и редактирования панелей, бинов и размерных стандартов в программе, а также их экспорта и импорта.
Область применения:
ДНК-идентификация и установление биологического родства в криминалистике, судебной медицине и других областях.
Наработка массивов данных для популяционных исследований научно-исследовательскими группами.
Молекулярное маркирование и генетическая паспортизация в селекции и биотехнологии.
Ветеринария и другие направления, базирующие свою работу на анализе коротких тандемных повторов ДНК.
Helicon Genmap имеет свидетельство о государственной регистрации программы для ЭВМ № 2024615693 от 12.03.2024.
Зарегистрировано в Минцифры России. Реестровая запись № 22458 от 14.05.2024.
Хеликон
Москва
Произведено в: Москва
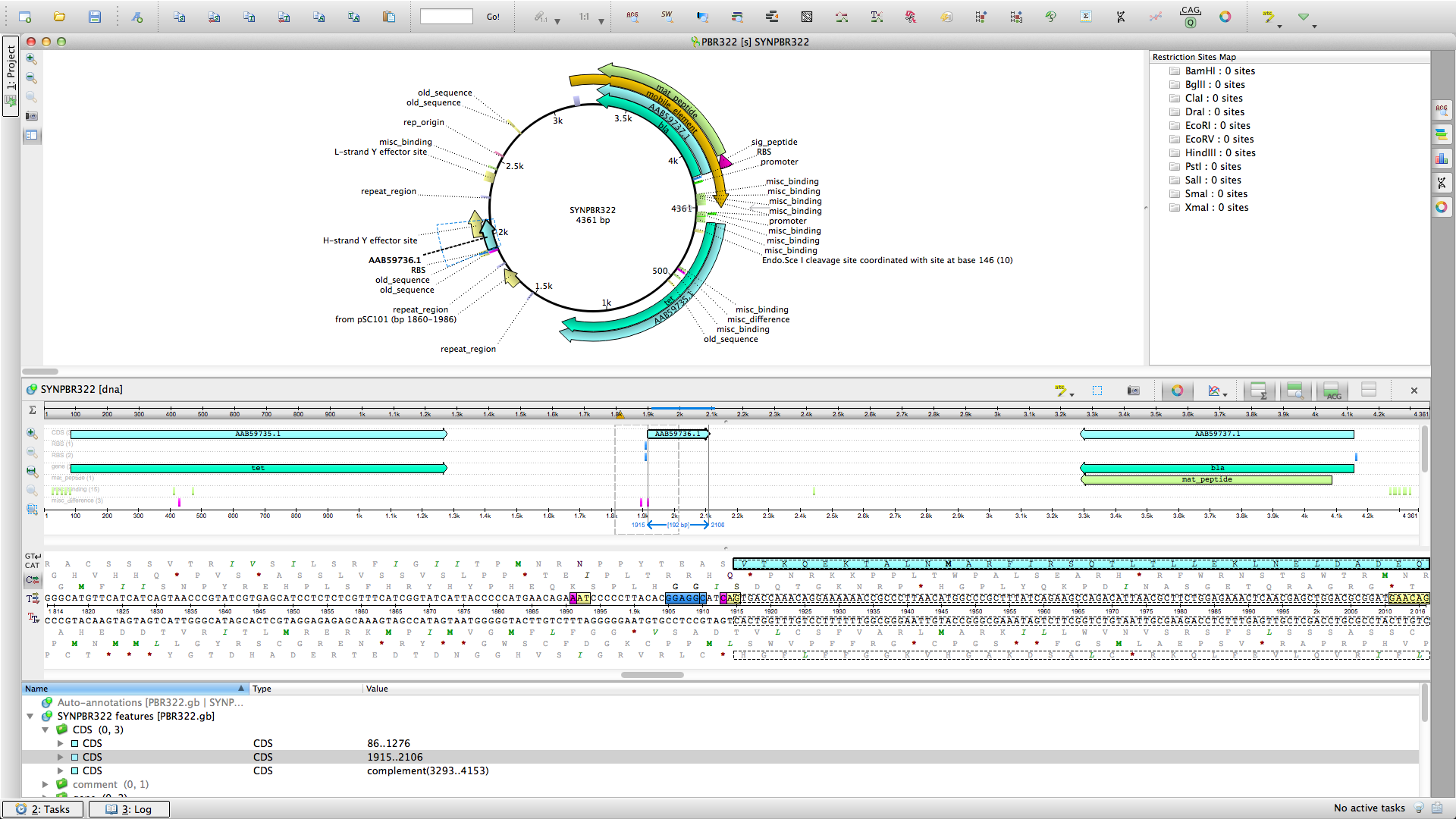
UGENE - открытая платформа биоинформатики
Unipro UGENE предоставляет множество методов обработки генетических и белковых последовательностей. Это — анализ, аннотирование, сравнение, поиск, выравнивание, моделирование, комплексные вычисления. Разнообразная визуализация, навигация и масштабирование, поддержка множества генетических форматов данных , оптимизация алгоритмов, возможности редактирования и графического построения различных вычислительных схем существенно ускоряют работу. UGENE отлично работает на Windows, Mac OS, Linux и прост в установке и использовании. Обладает интерфейсом как на русском, так и на английском языке.
Редактирование и аннотирование последовательностей
Поиск функциональных элементов и гомологов
Выравнивание и филогения
Клонирование in silico/ПЦР in silico
Работа с хроматограммами
Обработка данных NGS — сборка и визуализация генома, транскриптома, ChiP-Seq
Поиск геномных вариаций
Поиск в удаленных базах данных
Дизайнер вычислительных схем для многошагового анализа с десятками готовых схем
Произведено в: Новосибирск
Веб-сервис GeneCut
Основные функции программы включают:
Обратная трансляция аминокислотной последовательности с оптимизацией кодонного состава под определённый организм.
Оптимизация кодонного состава входной нуклеотидной последовательности.
Исключение определённых сайтов рестрикции и/или сплайсинга и/или пользовательских мотивов из нуклеотидной последовательности.
Разбиение нуклеотидной последовательности на олигонуклеотиды для последующей сборки одним из методов:
Polymerase Cycling Assembly (PCA),
Thermodynamically Balanced Inside-Out (TBIO).
Разбиение нуклеотидной последовательности на длинные блоки для последующей сборки одним из методов:
Gibson assembly,
Overlap extension PCR.
Клонирование одного или нескольких фрагментов в плазмиду одним из методов:
Gibson assembly,
Gateway.
Произведено в: Новосибирск

SNP TOOLBOX
Мощная интегрированная база данных, собранная по наиболее известным мировым источникам молекулярно-биологических данных. Промышленная бета-версия находится на стадии тестирования..
Инструмент помогает:
быстро получать исчерпывающую информацию по каждой вариации (гены, риски, болезни)
выбирать интересующие вариации, с помощью программных фильтров (по болезням, по положению в генах/хромосомах, по повреждающему эффекту, по встречаемости, а также неизвестные)
детально визуализировать интересующие вариации
Бета-версия доступна по запросу на snp-dev[ ]unipro.ru
Произведено в: Новосибирск
SNP TOOLBOX - геномные вариации
Наш инструмент помогает:
быстро получать исчерпывающую информацию по каждой вариации (гены, риски, болезни)
выбирать интересующие вариации, с помощью программных фильтров (по болезням, по положению в генах/хромосомах, по повреждающему эффекту, по встречаемости, а также неизвестные)
детально визуализировать интересующие вариации
Вариации можно загружать в любом доступном формате (vcg, gvf, …) полученные в любом исследовании, будь то секвенирование, PCR диагностика или микрочиповый анализ. Каждая загруженная вариация сравнивается с объединенной базой данных, собранной из курируемых открытых мировых источников (UCSC, OMIM, GAD,…) , и сводная информация выдается единым блоком. Можно отбирать из загруженных вариаций разные списки, удовлетворяющие критериям поиска. Отчет можно распечатать как по списку вариаций, так и по каждой в отдельности.
Произведено в: Новосибирск

Gency (Genotype frequency)
Программа предназначена для расчёта вероятности случайного совпадения комбинированных генотипов для анализируемых биологических образцов при ДНК исследованиях и может быть использована в экспертной практике судебно-медицинских, криминалистических, клинико-диагностических, гистологических и других лабораторий, занимающихся идентификацией происхождения биологических образцов (идентификацией личности).
Программа обеспечивает выполнение следующих функций:
- выбор эталонной популяции для расчётов;
- ввод генотипов для одного обследованного лица (объекта) по отдельным локусам;
- расчёт популяционной частоты встречаемости выявленного генотипа по каждому локусу;
- расчёт популяционной частоты встречаемости комбинированного генотипа по нескольким выбранным локусам;
- сохранение результатов работы в отдельном файле;
- возможно изменение конфигурации служебных файлов по усмотрению пользователя, в том числе создание опорных (эталонных) частот аллелей для любой локальной популяции.
ФГУП «ГосНИИгенетика»
Москва
Произведено в: Москва